Sequencing-based spatial gene expression
Transcriptomics Spatialhttps://www.nature.com/articles/s41592-024-02325-3
Sequencing-based approaches, such as Visium or Slide-Seq, are based on in situ transcript capture followed by ex-situ sequencing, with the addition of barcodes to link each captured molecule to its original spatial position. Those approaches allow whole-transcriptome gene expression profiling, at the cost of a low resolution dependant of the size of capture spots or barcoded beads.
Production systems

10xGenomics Visium
Spatial transcriptomics approaches conserve the localization of transcript expression in tissue. This spatial information is crucial for contextualizing a transcriptomic phenotype observed at histological level and deducing its function, or for comparing pathology-induced expression variations within the same tissue. 10X Genomics Visium is a spatial genomics platform that captures whole transcriptome gene expression data within FFPE or fresh frozen tissue sections, providing spatial context to transcriptomic information. It involves depositing a histological section about ten micrometers thick on a 6.5 mm square slide, gridded by 5,000 “spots” containing specific positional barcodes. These spots measure 55 μm and their centers are separated from each other by 100μm. Tissue cells are first permeabilized so that their RNAs are captured by the underlying spots and then retrotranscribed. The resulting cDNAs are then pooled and prepared for Illumina sequencing.
See more about the technologyAssociated pipelines
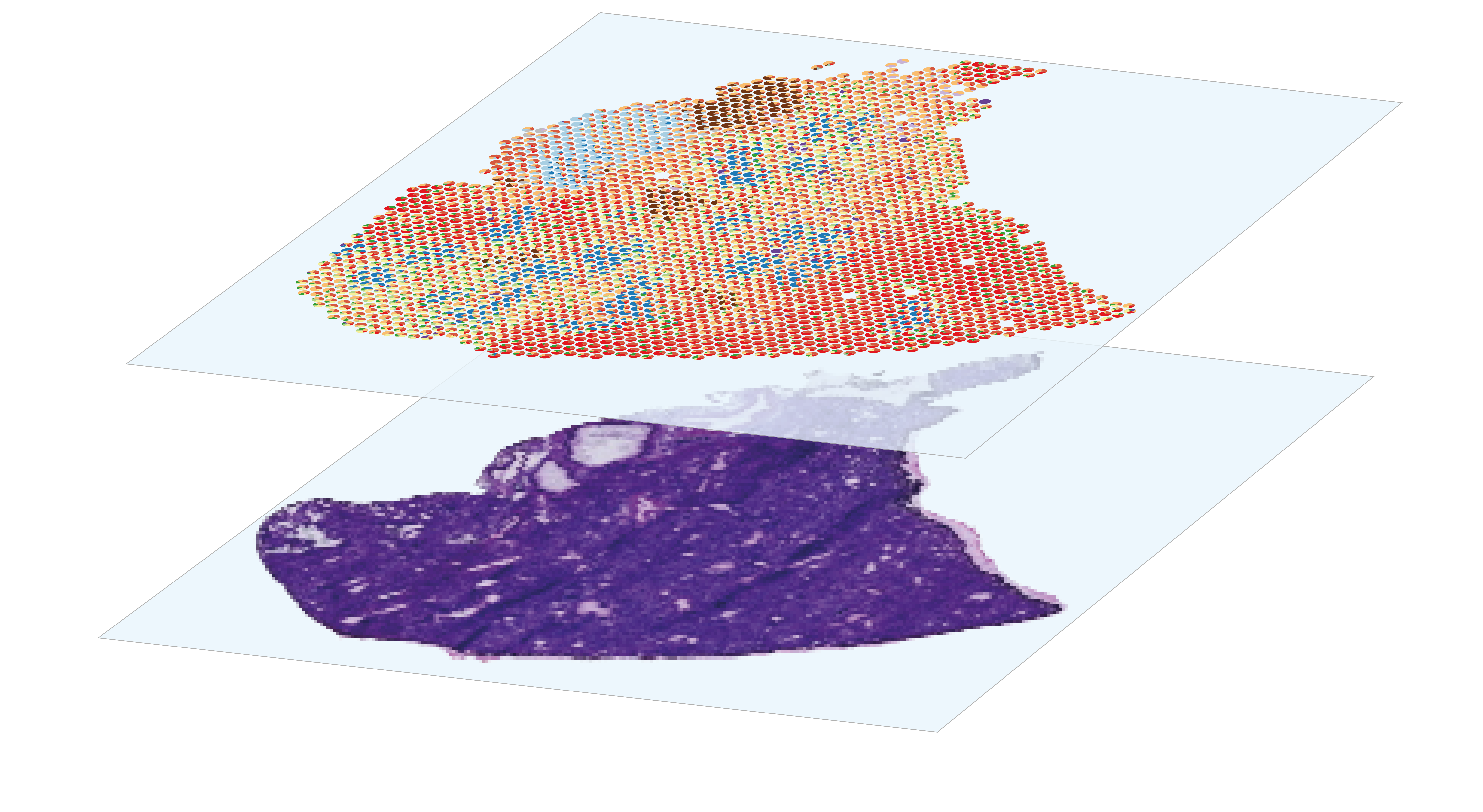
sequencing-based spatial transcriptomics
The analysis of spatial gene expression data generated by the Visium system begins with preprocessing the raw data with space ranger. Briefly, it consists in performing demultiplexing and quality control to filter out low-quality reads, aligning the remaining reads to a reference before counting gene expression from a single capture area. Once the matrix of gene counts per spot has been obtained, the aim is to identify the cell types represented in each spot. Although Visium technology avoids the dissociation step, its resolution is too low to perform single-cell characterization, as each 55µm diameter spot contains several cells. This implies that each spot is a combination of the transcriptome of potentially heterogeneous populations, which can be estimated using deconvolution methods, leveraging or not on a single-cell RNA-seq expression profils reference. After deconvolution, we can compare the distribution of cell types by histological region within the same section, or between sections from different experimental conditions. Combined with the spatial gene expression patterns, spots annotation can be used for Niche analysis, in order to characterize the interactions and relationships between cells and their microenvironment within tissues.
Go to pipeline documentation